From README.md
CoMD is a reference implementation of typical classical molecular dynamics algorithms and workloads. It is created and maintained by ExMatEx: Exascale Co-Design Center for Materials in Extreme Environments (exmatex.org). The code is intended to serve as a vehicle for co-design by allowing others to extend and/or reimplement it as needed to test performance of new architectures, programming models, etc.
Analysis
Parameters
Compiler = icc (ICC) 18.0.1 20171018
Build_Flags = -std=c99 -g -O3 -march=native -qopenmp -DDOUBLE
Run_Parameters = -x 128 -y 128 -z 50
Scaling
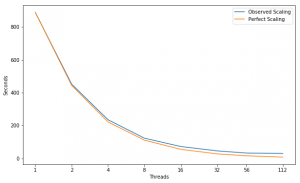
Performance Improvement
| Threads | |||||||
|---|---|---|---|---|---|---|---|
| Speed Up | 1.97X | 1.92X | 1.91X | 1.70X | 1.58X | 1.40X | 1.09X |
Hit Locations

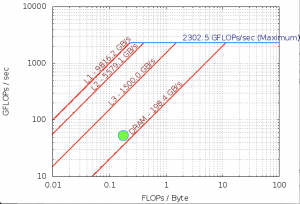
FLOPS
| Double Precision | ||||||
|---|---|---|---|---|---|---|
| PMU | 1.750e+12 | 9.130e+06 | 8.810e+07 | 0.000e+00 | 1.750e+12 | 5.327e+01 |
| SDE | 1.843e+12 | 7.252e+06 | 7.986e+07 | 0.000e+00 | 1.843e+12 | 5.609e+01 |
Intel Software Development Emulator
| Intel SDE | |
|---|---|
| Arithmetric Intensity | 0.177 |
| FLOPS per Inst | 0.56 |
| FLOPS per FP Inst | 1.0 |
| Bytes per Load Inst | 7.95 |
| Bytes per Store Inst | 7.89 |
@NOTE: Not Vectorized
Roofline – Intel(R) Xeon(R) Platinum 8180M CPU
112 Threads – 56 – Cores 3200.0 Mhz
UOPS Executed
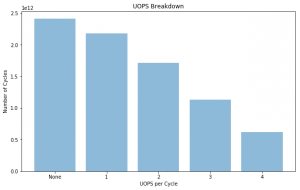
@NOTE: Getting use out of OOO core pipeline
Experiment Aggregate Metrics
| 1 (100.0%) | 1.46 | 1.03 | 1.02 | 0.14% | 49.35% | 80.79% | 0.50% | 2.01% | 2.17% |
| 56 (100.0%) | 0.85 | 0.59 | 0.58 | 0.12% | 47.65% | 82.64% | 0.26% | 2.48% | 7.13% |
| 112 (100.0%) | 1.07 | 0.38 | 0.37 | 0.15% | 38.68% | 76.07% | 0.43% | 3.22% | 9.09% |
ljForce
145 int ljForce(SimFlat* s)
146 {
147 LjPotential* pot = (LjPotential *) s->pot;
148 real_t sigma = pot->sigma;
149 real_t epsilon = pot->epsilon;
150 real_t rCut = pot->cutoff;
151 real_t rCut2 = rCut*rCut;
152
153 // zero forces and energy
154 real_t ePot = 0.0;
155 s->ePotential = 0.0;
156 int fSize = s->boxes->nTotalBoxes*MAXATOMS;
157 #pragma omp parallel for
158 for (int ii=0; ii<fSize; ++ii)
159 {
160 zeroReal3(s->atoms->f[ii]);
161 s->atoms->U[ii] = 0.;
162 }
163
164 real_t s6 = sigma*sigma*sigma*sigma*sigma*sigma;
165
166 real_t rCut6 = s6 / (rCut2*rCut2*rCut2);
167 real_t eShift = POT_SHIFT * rCut6 * (rCut6 - 1.0);
168
169 int nNbrBoxes = 27;
170
| 1 (95.4%) | 1.47 | 1.04 | 1.04 | 0.08% | 34.51% | 47.56% | 0.30% | 0.82% | 0.87% |
| 56 (58.3%) | 1.20 | 0.84 | 0.84 | 0.07% | 33.20% | 66.07% | 0.21% | 1.38% | 4.06% |
| 112 (49.0%) | 1.64 | 0.58 | 0.58 | 0.11% | 34.28% | 57.56% | 0.44% | 2.94% | 8.06% |
171 // loop over local boxes
172 #pragma omp parallel for reduction(+:ePot)
173 for (int iBox=0; iBoxboxes->nLocalBoxes; iBox++)
174 {
175 int nIBox = s->boxes->nAtoms[iBox];
176
177 // loop over neighbors of iBox
178 for (int jTmp=0; jTmp<nNbrBoxes; jTmp++)
179 {
180 int jBox = s->boxes->nbrBoxes[iBox][jTmp];
181
182 assert(jBox>=0);
183
184 int nJBox = s->boxes->nAtoms[jBox];
185
186 // loop over atoms in iBox
187 for (int iOff=MAXATOMS*iBox; iOff<(iBox*MAXATOMS+nIBox); iOff++)
188 {
189
190 // loop over atoms in jBox
191 for (int jOff=jBox*MAXATOMS; jOff<(jBox*MAXATOMS+nJBox); jOff++)
192 {
193 real3 dr;
194 real_t r2 = 0.0;
195 for (int m=0; m<3; m++)
196 {
197 dr[m] = s->atoms->r[iOff][m]-s->atoms->r[jOff][m];
198 r2+=dr[m]*dr[m];
199 }
200
201 if ( r2 <= rCut2 && r2 > 0.0)
202 {
203
204 // Important note:
205 // from this point on r actually refers to 1.0/r
206 r2 = 1.0/r2;
207 real_t r6 = s6 * (r2*r2*r2);
208 real_t eLocal = r6 * (r6 - 1.0) - eShift;
209 s->atoms->U[iOff] += 0.5*eLocal;
210 ePot += 0.5*eLocal;
211
212 // different formulation to avoid sqrt computation
213 real_t fr = - 4.0*epsilon*r6*r2*(12.0*r6 - 6.0);
214 for (int m=0; m<3; m++)
215 {
216 s->atoms->f[iOff][m] -= dr[m]*fr;
217 }
218 }
219 } // loop over atoms in jBox
220 } // loop over atoms in iBox
221 } // loop over neighbor boxes
222 } // loop over local boxes in system
223
224 ePot = ePot*4.0*epsilon;
225 s->ePotential = ePot;
226
227 return 0;
228 }